Kombinierter Immundefekt (CID)

Kinder mit einem Kombinierten Immundefekt (combined immunodeficiency = CID) leiden an seltenen angeborene Erkrankungen des Abwehrsystems (Immunsystems). Im Gegensatz zum SCID (Schwerer Kombinierter Immundefekt) sind jedoch Restfunktionen des Abwehrsystems erhalten und damit auch ein Teilschutz gegenüber Krankheitserregern und Parasiten erhalten. Der Begriff „kombiniert“ wird verwendet, da sowohl die Abwehrzellen als auch die Bildung von sogenannten Antikörpern gegen Erreger betroffen sein können.
Da bei den kombinierten Immundefekten beide Anteile des Immunsystems beeinträchtigt sind, ist das klinische Bild variabel und von Fall zu Fall unterschiedlich. „Normale“ Keime, die Menschen mit gesundem Abwehrsystem nicht gefährden, können bei Kindern mit CID zu schweren Infektionen führen.
Darüber hinaus treten Infektionen mit Keimen auf, die sonst nur sehr selten zur Erkrankung führen. So können Erreger wie Pneumocystis jirovecii, Aspergillus, Cytomegalieviren oder Cryptosproridien bei Kindern mit CID zu schweren Lebererkrankungen, Lungenentzündungen und schweren Durchfällen führen. Kinderkrankheiten wie Windpocken, Röteln oder Herpes-Infektionen oder sonst unkomplizierte Pilzerkrankungen (Windelsoor), können bei Kindern mit CID lebensbedrohlich verlaufen.
Die Fehlsteuerung kann sich auch dadurch äußern, dass sich das Abwehrsystem gegen den eigenen Körper richtet. Es kommt dann zu Autoimmunerkrankungen, die vor allem gegen Blutzellen, manchmal auch gegen Haut, Gelenke, Augen oder die Schilddrüse gerichtet sind. Bei einigen Patienten schwellen Lymphdrüsen, Leber oder Milz an. Ekzeme, chronische Darmentzündung, unklare Fieberschüben, Granulombildung (Knötchenbildung in der Haut oder in inneren Organen) können Ausdruck einer Fehlsteuerung des Immunsystems sein. Aber auch Entzündungsgeschehen und ein erhöhtes Krebsrisiko gehören dazu.
Im Vergleich zu anderen Kindern werden Kinder mit kombiniertem Immundefekt häufiger krank, Infekte verlaufen schwerwiegender und die betroffenen Kinder brauchen länger, um sich zu erholen. Im Vordergrund stehen meist vor allem bakterielle Infektionen der Atemwege (Mittelohrentzündungen, Nebenhöhlenentzündungen, Entzündungen der Bronchien und der Lunge). Auch Durchfallerkrankungen sind häufig. Oft sind wiederholte und längere Behandlungen mit Antibiotika nötig.
Die Bildung von Antikörpern gegen eigene Blutzellen kann dazu führen, dass im Körper deutlich zu wenige Blutzellen vorhanden sind (Autoimmunzytopenie). Dies führt zu Blässe, Kopfschmerzen und Leistungsknick durch Blutarmut (Anämie), Störung der Blutstillung durch zu wenige Blutplättchen (Thrombopenie) und/oder zu Infektanfälligkeit durch Verminderung der weißen Blutzellen (Neutropenie).
Durch ihr variables Erscheinungsbild äußert sich die CID-Erkrankung bei jedem Patienten anders. Das Ausmaß der Infektneigung oder der Autoimmunerscheinungen kann sehr unterschiedlich sein. Auch der Verlauf der Erkrankung ist von Patient zu Patient sehr verschieden. Der Beginn der Krankheitserscheinungen kann im Kindesalter oder auch erst im frühen Erwachsenenalter liegen.
Der Begriff CID ist von CVID (common variable immunodeficiency) und von SCID (severe combined immunodefien- cy ) abzugrenzen. Bei der CVID-Erkrankung steht der Antikörpermangel im Vordergrund, die Abwehrzellen selbst sind in der Regel nur wenig beeinträchtigt. Die begriffliche Abgrenzung ist allerdings nicht ganz genau, der Übergang zum CID ist fließend. Beim SCID steht die Störung der Abwehrzellen im Vordergrund. Da für eine ausreichende Antikörperbildung aber auch zelluläre Funktionen notwendig sind, liegt beim SCID immer auch ein sekundärer Antikörpermangel vor. Der Verlauf der SCID-Erkrankung ist so schwer, dass es bereits im Säuglingsalter zu schweren Infektionen kommt und betroffene Kinder ohne Knochenmarkstransplantation vor dem zweiten Lebensjahr versterben (siehe Patientenbroschüre SCID).
Beim CID sind Restfunktionen der Abwehr erhalten und schützen den Patienten. Im Vordergrund steht ebenfalls die Störung der zellulären Abwehr, allerdings ist sie nicht so schwer wie beim SCID. Die Antikörperbildung ist in variablem Ausmaß auch betroffen. Patienten mit CID können verhältnismäßig gesund sein und zeigen erst später, manchmal auch erst im Erwachsenenalter, Symptome die auf einen Immundefekt hinweisen.
CID ist eine seltene Erkrankung. Zählt man alle Unterformen zusammen, tritt CID in Deutschland ungefähr bei 1 von 10.000 Personen auf.
Das gesunde Abwehrsystem ermög-licht dem Körper Krankheitserreger zu erkennen und abzuwehren. Zellen des Abwehrsystems (lymphatische Vorläuferzellen) reifen wie auch die anderen Blutzellen, zunächst im Knochenmark heran und durchlaufen dann verschiedene Stadien, um sich zu funktionstüchtigen Abwehrzellen zu entwickeln.
Eine wichtige Station der Zellreifung nach dem Knochenmark ist der Thymus, ein Organ hinter dem Brustbein. Im Thymus werden ein Teil der Lymphozyten, die nach dem Ort ihrer Prägung T-Zellen genannt werden, für ihre spätere Funktion im Abwehrsystem „geschult“. Einerseits vermehren sie sich im Thymus, andererseits „lernen“ sie, zwischen körpereigenen Bestandteilen (Selbst-Antigenen) und Erregern (Fremdantigenen) zu unterscheiden. Erst wenn die T-Zellen die „Thymusschule“ durchlaufen haben, sind sie in der Lage, ihre spezifischen Aufgaben im Immunsystem zu übernehmen.
Wenn die T-Zellen den Thymus verlassen, wandern sie über Blut und Lymphsystem durch den Körper. Nach Infektion mit einem Erreger werden sie aktiviert und bilden wichtige Entzündungs- und Botenstoffe, die einerseits auf die Erreger wirken und andererseits regulierende Wirkung auf andere Abwehrzellen haben. Am Ende einer erfolgreichen Immunantwort kommen die T-Zellen wieder zur Ruhe, bilden aber ein „Gedächtnis“ aus, d.h. sie können bei einer erneuten Auseinandersetzung mit dem gleichen Erreger sehr viel schneller und effektiver reagieren.
Kombinierte Immundefekte sind vererbte Erkrankungen, die in erster Linie zu einer Störung der Entwicklung, Reifung oder der Aktivierung der T-Zellen führen. Kombinierter Immundefekt ist ein zusammenfassender Begriff, hinter dem sich eine Vielzahl von verschiedenen Erkrankungen verbirgt. Die Schwere des kombinierten Defekts ist davon abhängig, welcher Vorgang genau in dem Lebenszyklus der T-Zellen gestört ist. Eine komplette Störung der Ausreifung von T-Zellen führt zum schweren kombinierten Immundefekt (SCID). Ist die Störung aber inkomplett und lässt noch eine gewisse Reifung von T-Zellen zu, ist das klinische Bild milder und der Beginn der Krankheitserscheinungen später. Eine Verminderung von T-Zellen ist charakteristisch für kombinierte Immundefekte, manchmal haben die Patienten aber auch normale Zahlen an T- Zellen, die jedoch nicht richtig aktiviert werden können. Andere Immunzellen sind in sehr variablem Ausmaß in ihrer Zahl und Funktion beeinträchtigt.
Die Ursache der Entwicklungs- oder Funktionsstörung von T-Zellen sind Gendefekte. Das klinische Bild eines kombinierten Immundefekts kann durch eine Vielzahl verschiedener Gendefekte (über 40 Gene) hervorgerufen werden. Hinzu kommt, dass bisher nicht alle Gene, die für die komplizierte Entwicklung und Funktion von T-Zellen von Bedeutung sind, bekannt sind. Dies führt dazu, dass mit den heutigen Methoden nicht bei allen Patienten mit CID auch ein ursächlicher Gendefekt gefunden werden kann. Mit Hilfe der Krankheitserscheinungen und einer Reihe von Untersuchungen des Immunsystems ist es aber in vielen Fällen möglich, die Erkrankung genauer zu charakterisieren und auch in Abwesenheit einer genetischen Diagnose ein klares Therapiekonzept zu erstellen.
Gendefekte sind Erbkrankheiten. Wenn eine Erbkrankheit vorliegt, bedeutet das, dass der Patient von Mutter und/ oder Vater das fehlerhafte Gen geerbt hat. Jeder Mensch besitzt von jedem Gen zwei Stück, eines vom Vater und eines von der Mutter. Für die meisten Erbkrankheiten ist es nötig, dass beide Gene fehlerhaft sind, da ein gesundes Gen in der Regel ausreicht. Bei den kombinierten Immundefekten gibt es verschiedene Formen der Vererbung.
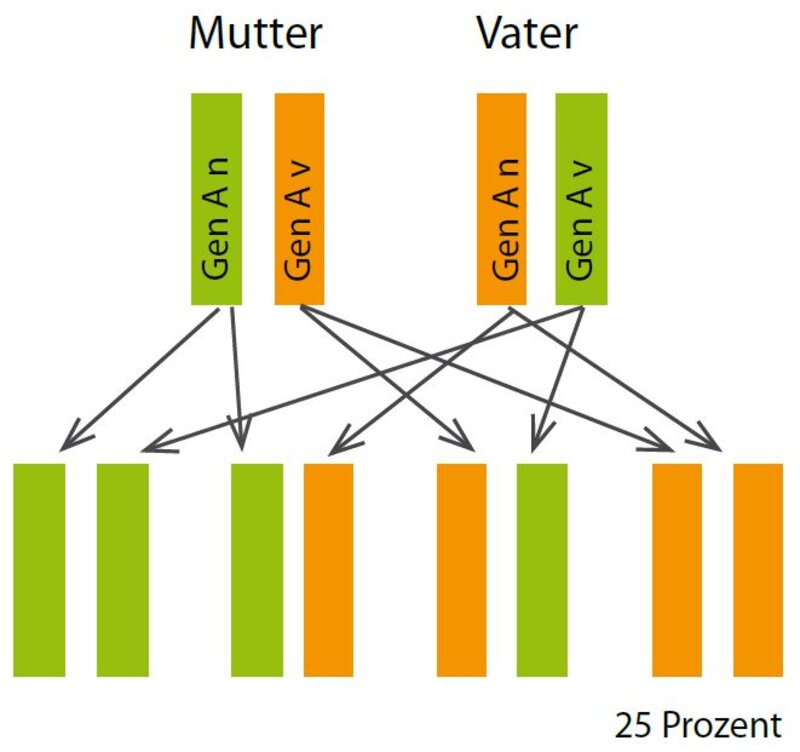
Beim autosomal-rezessiven Erbgang sind beide Elternteile gesund. Das normale Gen (Gen An, grün) setzt sich gegenüber dem veränderten Gen (Gen Av) durch. Jedes Kind bekommt von jedem Elternteil eine Genvariante vererbt. Die Nachkommen, die von beiden Elternteilen das veränderte Gen erben, erkranken. Die gesunden Geschwister, die ein verändertes Gen tragen, können dieses wieder an ihre Kinder weitergeben.
Autosomal-rezessiv
Das bedeutet, dass beide Elternteile klinisch gesunde Träger des defekten Gens sind, d.h. beide haben neben dem krankmachenden auch ein gesundes Gen, das ausreicht, sie vor der Erkrankung zu schützen. Für die Nachkommen besteht eine Erkrankungswahrscheinlichkeit von 25 Prozent. Die Hälfte der Kinder werden wie ihre Eltern klinisch gesunde Genträger, können aber das Gen an ihre Kinder weitervererben und 25 Prozent sind ganz gesund. Die Vererbung ist hier geschlechtsunabhängig.
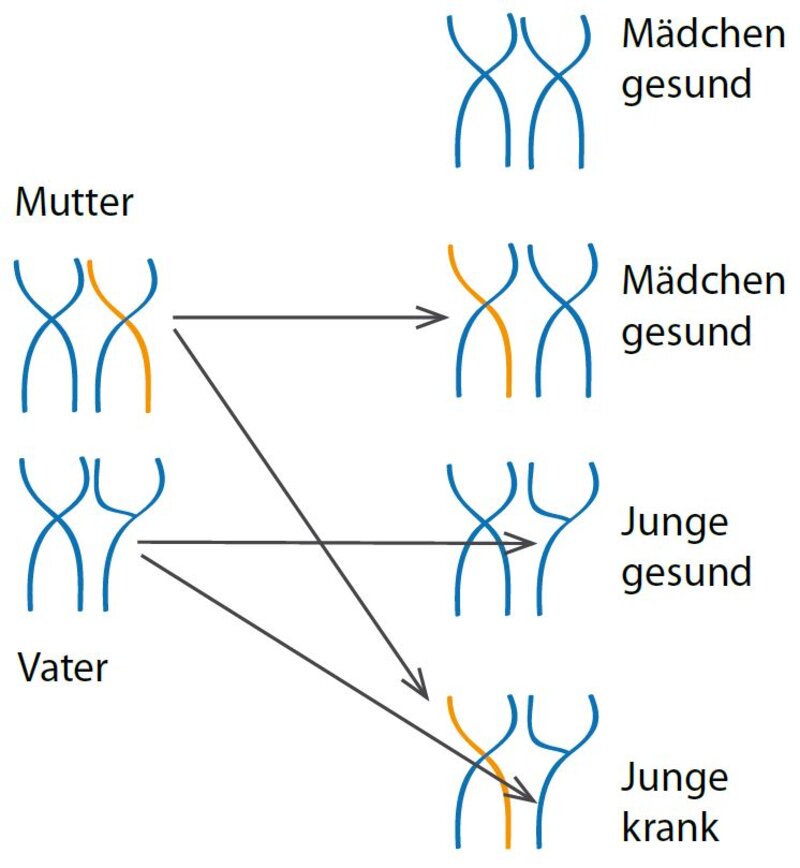
Bei der X-chromosomalen Vererbung liegt der Defekt auf dem Geschlechtschromosom X. Da Mädchen jeweils zwei X-Chromosomen besitzen erkranken sie nicht, da das gesunde Chromosom den Defekt ausgleicht. Jungen erkranken dann, wenn sie das kranke Gen von der Mutter erben.
X-chromosomal
Hier ist meist die Mutter die klinisch gesunde Trägerin des kranken Gens, kann das aber durch ein gesundes Gen auf ihrem anderen X-Chromosom ausgleichen. Jungen haben nur ein X-Chromosom, das sie als eines von zwei mütterlichen X-Chromosomen von der Mutter erben. Wenn sie das X-Chromosom mit dem kranken Gen erben, tritt die Krankheit auf, da sie nicht mit einem zweiten gesunden Gen ausgeglichen werden kann. Bei diesem Erbgang ist die Hälfte aller Jungen von der Krankheit betroffen, während die Mädchen alle gesund sind. Die Hälfte der Mädchen kann allerdings die Krankheit wieder an ihre eigenen männlichen Kinder übertragen.
In der Regel fallen Patienten mit einem kombinierten Immundefekt bereits im Kindesalter durch immer wiederkehrende Infektionen, Autoimmunerkrankungen (z.B. durch Antikörper vermittelte Blutarmut oder Blutungsneigung), unklare Ekzeme oder chronische Darmentzündung auf. Diese Probleme führen zu gehäuften Arztbesuchen. Manchmal ist der erste Hinweis auf eine CID Erkrankung auch eine schwer verlaufende Infektion, die zu einer stationären Aufnahme führt. Diese Infektionen können so schwer sein, das unter Umständen ein Aufenthalt auf einer Intensivstation und eine intensive Therapie erforderlich sind. Oft sind auch Wachstum und Entwicklung durch die wiederholten Infektionen gestört. Wenn der Verdacht eines kombinierten Immundefektes besteht, sollten die Kinder so schnell wie möglich in ein auf Immundefekte spezialisiertes Zentrum überwiesen werden, da eine rasche Diagnosestellung für die Einleitung der richtigen Behandlung wichtig ist.
Besteht der Verdacht auf einen Immundefekt, ist zunächst eine genaue Erhebung der Anamnese (Krankheitsgeschichte) unter Mitbeurteilung der Familiengeschichte (wie z.B. Blutsverwandtschaft, bekannter Immundefekt in der Familiengeschichte) sowie eine sorgfältige körperliche Untersuchung von Bedeutung. Besondere Beachtung finden bei der körperlichen Untersuchung die Haut (bei manchen Patienten finden sich Neurodermitis-ähnliche Hautausschläge), die Lunge (Bronchitis und Lungenentzündung sind beim CID häufig), Leber und Milz (die beim CID vergrößert sein können) sowie Lymphknoten und Mandeln, die ebenfalls vergrößert sein können. Manche kombinierte Immundefekte sind syndromale Erkrankungen, bei denen kleinere Fehlbildungen (Gesichtsform, Ohrmuscheln, Zahnschmelz oder Zahnform) Hinweise auf die genaue Erkrankung geben können. Gewicht, Größe und Kopfumfang werden ermittelt, um festzustellen ob sich das Kind altersentsprechend entwickelt hat.
Zusätzlich zu der Erhebung der Krankengeschichte und der körperlichen Untersuchung sind mehrere Bluttests erforderlich. Die erste wichtige Labordiagnostik ist hierbei die sorgfältige Beurteilung des Blutbildes, wobei die Anzahl der Lymphozyten (einer Untergruppe der weißen Blutkörperchen) von großer Bedeutung ist. Eine geringe Anzahl von Lymphozyten ist ein wichtiger Hinweis für kombinierte Immundefekte. Darüber hinaus werden die Antikörperspiegel gemessen, die oft erniedrigt sind. Falls das Kind schon geimpft worden ist, kann außerdem geprüft werden, ob es in der Lage ist, gegen die Impfstoffe spezifische Antikörper zu bilden. Sind die Lymphozyten erniedrigt, werden die verschiedenen Untergruppen bestimmt (T-Zellen, B-Zellen und NK- Zellen) und deren Funktion im Reagenzglas untersucht. Das Muster der Störungen in den unterschiedlichen Zelltypen lässt Rückschlüsse auf die Art der CID-Erkrankung zu.
Neben den Blutuntersuchungen ist zur genaueren Einordnung häufig eine Hautbiopsie (Entnahme eines kleinen Hautstückchens) oder aber auch die Untersuchung von Gewebe der Lymphknoten, der Darmschleimhaut oder des Knochenmarks erforderlich. Mit Hilfe von bildgebenden Verfahren wie Röntgen und Ultraschall lässt sich fest- stellen, ob Organvergrößerungen oder -veränderungen vorliegen, z.B. Knötchen in der Lunge oder in der Leber. Je nach klinischer Situation werden verschiedene weitere Untersuchungen wie z.B. Urin-, Stuhluntersuchungen und evtl. auch eine Lungenspiegelung (Bronchoskopie) durchgeführt. Bei einer Bronchoskopie kann die Lunge mit Hilfe eines speziellen Gerätes direkt eingesehen und Sekret entnommen werden, welches dann auf evtl. vorhandene Keime untersucht werden kann. Auch Darmspiegelungen können erforderlich sein, um chronische Durchfälle und Darmentzündungen besser einordnen zu können. Sind solche Untersuchungen erforderlich, werden Sie ausführlich aufgeklärt und haben genügend Zeit, Fragen zu stellen.
In einem Teil der Fälle von CID kann die klinische Verdachtsdiagnose im weiteren Verlauf durch einen Gentest gesichert werden. Diese Untersuchungen dauern aber manchmal mehrere Monate, so dass mit der Entscheidung zur Behandlung nicht immer darauf gewartet werden sollte. Patienten mit CID sollten immer in einem darauf spezialisierten Zentrum behandelt werden.
Die Behandlung der CID Erkrankung richtet sich nach der Form und Schwere der Erkrankung. Die Behandlungsprinzipien sind: Behandlung bereits vorliegender Infektionen, Vorbeugung und Verhindern von neuen Infektionen, Behandlung von Störungen der Immunregulation, Stärkung des Immunsystems durch Antikörperinfusionen und in vielen Fällen auch der Ersatz des Immunsystems durch Knochenmarks- transplantation.
Aufgrund der Störung des Immunsystems benötigt das Kind manchmal eine vorbeugende Gabe von bestimmten Medikamenten, um Infektionen abzuwehren. Eine vorbeugende Gabe von Antibiotika hilft, bakterielle Infektionen zu verhindern, antivirale Medikamente schützen gegen manche Viren und auch Medikamente gegen Pilze werden vorsorglich eingesetzt. Viele der Medikamente gibt es in Form von Säften, manchmal ist es aber auch nötig, dass ihr Kind die Medikamente über eine Infusion bzw. den zentralvenösen Katheter bekommt. Leider reichen diese Medikamente oft nicht aus, um Infektionen vollständig zu verhindern.
Impfungen sind bei Patienten mit kombiniertem Immundefekt in der Regel nicht sinnvoll. Die fehlende Antwort auf Totimpfstoffe wird diagnostisch eingesetzt. Lebendimpfungen (z.B. Masern, Windpocken, Rotavirus) sollten nicht verabreicht werden.
Immunglobuline
Kinder mit CID können aufgrund der fehlerhaften Abwehrzellen meist nicht genügend oder nicht die richtigen Antikörper (Immunglobuline) bilden, um gegen Infektionen ankämpfen zu können. Daher müssen diese Antikörper in Form einer Infusion ersetzt werden. Es ist möglich die Antikörper über die Vene (alle vier Wochen) oder auch subkutan in das Unterhaufettgewebe (einmal pro Woche) zu geben. Da die Antikörper nach einer gewissen Zeit im Körper abgebaut werden, müssen die Infusionen regelmäßig wiederholt werden. Die Handhabung der Pumpe kann für den Heimgebrauch erlernt werden. Mehr Informationen zur Antikörperersatztherapie gibt es in einem separaten Merkblatt.
Immunsuppressiva
Bei Kindern, die eine Störung der Immunregulation zeigen (z.B. Ekzeme, Durchfälle, Milzvergrößerung, Autoimmunität) müssen manchmal Medikamente eingesetzt werden, die fehlgesteuerte, überschießende Immunreaktionen unterdrücken. Zu dieser Gruppe von Medikamenten gehören die sogenannten Immunsuppressiva. Das am häufigsten eingesetzte Medikament ist Cortison, das eine sehr gute und rasch einsetzende anti-entzündliche Wirkung hat. Bei längerer Anwendung kommt es meist zu unerwünschten Nebenwirkungen, so dass zusätzliche Cortison-sparende Medikamente zur Anwendung kommen, bei denen in der Regel der Wirkungseintritt etwas länger dauert. Der Einsatz dieser Immunsuppressiva bei CID muss vorsichtig erfolgen und erfordert einige Erfahrung, um das richtige Maß an Immunsuppression zu erzielen ohne dabei die Infektanfälligkeit wesentlich zu erhöhen. Manchmal wer- den auch Medikamente eingesetzt, die fehlregulierte Immunzellen abtöten (z.B. Rituximab, ein Medikament, das Antikörper-bildende B-Zellen zerstört). Mit diesem Medikament wird erreicht, dass die Bildung von krankmachenden Autoantikörpern gestoppt wird.
Milzentfernung
Bei manchen Patienten mit CID kommt es zu schwerer Auto-Antikörper-vermittelter Zytopenie, d.h. zu einer Zerstörung von Blutzellen oder Blutplättchen durch Antikörper. Häufig gelingt es, diese Zerstörung durch Gabe von Antikörpern, Cortison oder Immunsuppressiva zu stoppen. In manchen Fällen, ins- besondere bei Patienten mit deutlich vergrößerter Milz, reicht dies aber nicht immer aus. In solchen Fällen kann die Entfernung der Milz notwendig sein. Da eine Milzentfernung ein Infektanfälligkeitsrisiko darstellt, muss nach dem Eingriff eine Dauerprophylaxe mit Antibiotika durchgeführt werden, die aber in aller Regel gut vertragen wird.
Bluttransfusion
Es kann sein, dass ihr Kind eine Transfusion von Blutprodukten braucht. Hierzu gehören rote Blutkörperchen oder auch Blutplättchen. Falls ihr Kind Blut braucht, werden diese Blutproben speziell behandelt (bestrahlt), um alle Immunzellen aus der Konserve zu entfernen und somit das Risiko von Reaktionen zu verringern. Ebenso werden nur Konserven gegeben, die ganz besonders auf Krankheitserreger getestet wurden, da sonst harmlose Viren ein Risiko für Kinder mit Immundefekten darstellen können.
Knochenmarkstransplantation
Die Knochenmarkstransplantation (KMT) ist eine wichtige Therapieoption für Kinder mit CID, die es bei erfolgreicher Durchführung erlaubt, die Erkrankung vollständig zu heilen. Das Ziel dieser Therapie ist es, das kranke Abwehrsystem (Immunsystem) durch das Immunsystems eines gesunden Spenders zu ersetzen. Gesundes Knochenmark ist reich an Stammzellen. Stammzellen sind Zellen, die keine oder nur eine geringe Differenzierung aufweisen und somit in ihrer spätere Funktion im Organismus noch nicht festgelegt sind. Dadurch haben Stammzellen die Fähigkeit, sich zu verschiedenen Zelltypen entwickeln zu können, unter anderem zu Zellen des Immunsystems.
Wird ein geeigneter Spender gefunden, ist es möglich, dem betroffenen Kind gesundes Knochenmark mittels einer Infusion zu übertragen. Die Knochenmarkstransplantation ist keine Transplantation, wie man sie von an- deren Organen her kennt, sondern die im Knochenmark enthaltenen Stammzellen können über das Blut ihren Weg in das Knochenmark finden und dann dort beginnen, gesunde Blutzellen zu bilden.
Eine KMT birgt aber auch Risiken und es kann zu Komplikationen kommen. Meist sind die Komplikationen gut behandelbar, manche können aber auch einen lebensbedrohlichen Verlauf nehmen. Bevor es zu einer Therapieentscheidung kommt, wird ein Team von Spezialisten (Immunologen, Hämatologen) die Vorgehensweise, Risiken und Nutzen genau mit Ihnen besprechen und es wird ihnen genug Zeit gegeben, Fragen zu stellen und Unsicherheiten zu klären.
Um die KMT durchführen zu können, ist es wichtig einen geeigneten Spender zu finden. Daher wird bei Eltern und Geschwistern, manchmal auch bei weiteren Familienangehörigen Blut abgenommen, um die Merkmale bestimmen zu können die bei einer Transplantation übereinstimmen müssen. Wird innerhalb der Familie ein zu dem betroffenen Kind passender Spender gefunden (meist ein gesundes Geschwisterkind), so kommt diese Person als Spender in Frage. Ist innerhalb der Familie kein geeigneter Spender zu finden, wird eine weltweite Suche nach einem geeigneten Spender über einen Register veranlasst. Hiermit besteht heute eine sehr gute Chance, für die meisten Patienten einen Spender zu finden.
Ist ein geeigneter Spender gefunden, so beginnt die Therapievorbereitung für die KMT. In der Regel ist es notwendig, vor der KMT eine Chemotherapie durchzuführen, um das kindliche Immunsystem „herunterzuregulieren“ und damit das Risiko einer Abstoßung der transplantierten Stammzellen zu verringern. Wird ein sehr gut passender Spender innerhalb der Familie gefunden, ist bei CID eine vorhergehende Chemotherapie nicht immer erforderlich. Auch über die Risiken und Nebenwirkungen der Chemotherapie wird das Team der Hämatologie ausführlich mit Ihnen sprechen.
Nicht für alle Patienten mit CID ist die Therapie geeignet. In jedem Fall muss eine sorgfältige Risiko-Nutzen-Analyse erfolgen, in die der aktuelle Gesundheitsstatus, die spezielle Art der CID-Erkrankung und die bisher vorliegenden Erfahrungen, die Verfügbarkeit eines passenden Spenders und andere Faktoren eingehen. Letztlich wird gemeinsam mit der Familie eine Entscheidung getroffen, welches der richtige Weg ist.
Gentherapie
Eine neue Therapieform, die derzeit in Erprobung ist, ist die Gentherapie. Hier- bei werden bei dem erkrankten Kind Stammzellen entnommen, im Reagenzglas wird ihnen das defekte Gen in gesunder Form eingefügt und dann werden die Zellen dem Kind wieder zurückgegeben. Diese Behandlung hat den Vorteil, dass das Kind seine eigenen Zellen und nicht fremde Zellen bekommt. Damit sind die Risiken einer Abstoßung oder Unverträglichkeit deutlich geringer. Andererseits ist das Einschleusen eines neuen Gens in Stammzellen ein Vorgang, der eigene Risiken birgt. Der Eingriff kann dazu führen, dass die veränderten Zellen ihre Eigenschaften ändern und auch entarten (Krebs auslösen) können. Weltweit sind inzwischen über 50 Patienten mit der Gentherapie behandelt worden. Die meisten Behandlungen waren erfolgreich, so dass hiermit in der Zukunft eine echte Alternative zur KMT besteht, insbesondere, wenn kein passender Spender gefunden wird. Allerdings kam es in den frühen Studien bei einigen Patienten zur Entwicklung einer Leukämie. Es wird derzeit intensiv da- ran geforscht, sicherere Verfahren des Gentransfers zu entwickeln.
Die Prognose der CID Erkrankung ist letztendlich abhängig von der Form und dem Ausmaß der Störung. Ohne Behandlung kommt es immer wieder zu zum Teil schwer verlaufenden Infektionen und bei vielen Patienten zu gestörter Immunregulation. Mit Hilfe von Immunglobulinen, prophylaktischen Medikamente gegen Bakterien, Viren und Pilze sowie Immunsuppressiva können diese Krankheitszeichen vermindert, aber meist nicht vollständig vermieden werden. Häufig sind chronische Lungenerkrankungen ein Problem. Hier ist eine gute Lungenhygiene mit intensiver krankengymnastischer Behandlung ein ganz wichtiges Therapieelement. Langfristig heilen lässt sich die CID Erkrankung nur mit der Knochenmarkstransplantation oder Gentherapie.
Die Wirkung von Impfungen besteht darin, die Bildung von Antikörpern anzuregen. Bei Patienten mit CID kommt es jedoch aufgrund des T-Zelldefektes auch häufig zu einer Beeinträchtigung der B-Zellen und dadurch zu einer verminderten Antikörperbildung. Hier sind Impfungen dann nur von begrenztem Nutzen und können dem geschwächten Abwehrsystem auch schaden. Da- her sollten Impfungen mit Lebendimpfstoffen (Masern, Mumps, Röteln, BCG, Rotavirus) erst nach genauer Untersuchung der Lymphozyten durchgeführt werden. Schädliche Wirkungen durch Totimpfstoffe sind nicht zu befürchten. Manchmal kann eine Impfung mit einem Totimpfstoff wichtige Hinweise für die Einschätzung der CID-Erkrankung geben. Ob weitere Impfungen mit Totimpfstoffen sinnvoll sind, muss im Einzelfall mit dem behandelnden Arzt besprochen werden. Eine jährliche Impfung von engen Kontaktpersonen mit dem Influenza-Impfstoff ist sinnvoll.
Kinder mit CID-Erkrankung können in der Regel den Kindergarten und die Schule besuchen. In einigen Fällen sind allerdings besondere Vorsichtsmaßnahmen erforderlich, wie Vermeidung von Kontakt mit Infektionen. Es ist daher wichtig, dass sie mit ihrem behandelnden Arzt besprechen, worauf individuell geachtet werden sollte.
Stand
2012
Hinweis
Wir möchten mit unseren Patientenbroschüren gerne dazu beitragen, dass betroffene Patienten, Eltern und ihr Umfeld die Erkrankung und ihre Behandlung besser verstehen. Die Broschüren sind sorgfältig erstellt und beschreiben die Erkrankung und deren Behandlung. Auch wenn Sie viele Informationen in den Broschüren finden, können diese vorliegenden Informationen keinen Arztbesuch ersetzen.
Autor
Henrike Ritterbusch
+49 (0)761 270-45240
henrike.ritterbusch@uniklinik-freiburg.de
Wissenschaftliche Begleitung
Prof. Dr. Stephan Ehl
+49 (0)761 270-77300
stephan.ehl@uniklinik-freiburg.de
UNIVERSITÄTSKLINIKUM FREIBURG
Centrum für Chronische Immundefizienz
Mathildenstraße 1
79106 Freiburg
www.uniklinik-freiburg.de/cci
