Activated PI3 Kinase Delta Syndrom (APDS)

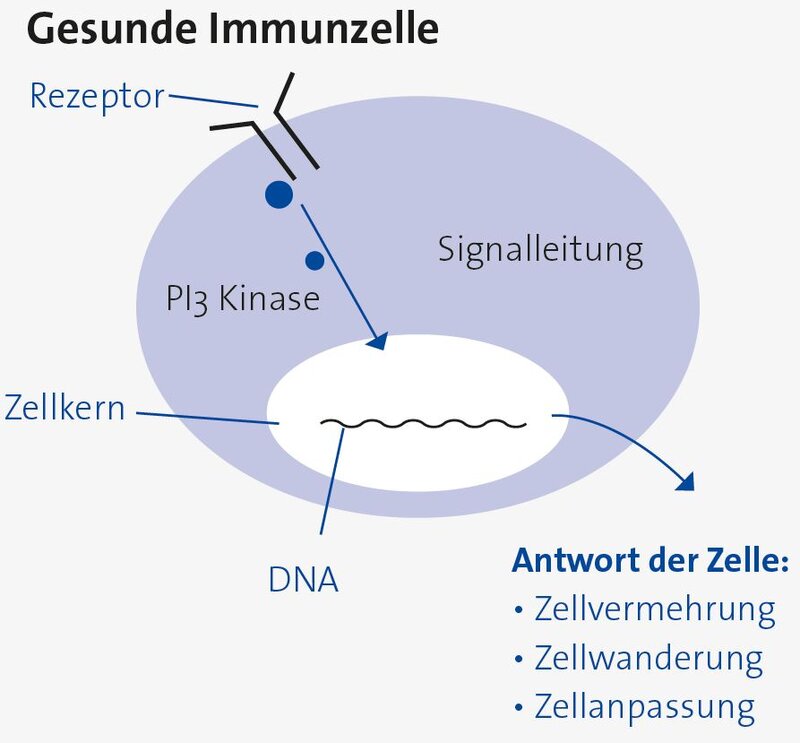
Beim Activated PI3 Kinase Delta Syndrom (APDS) handelt es sich um einen seltenen angeborenen Immundefekt, welchem ein genetischer Defekt zugrunde liegt. Die Erkrankung wird auch PASLI genannt (“p110 delta activating mutation causing senescent T cells, lymphadenopathy, and immunodeficiency”). Sie beruht auf einer Fehlsteuerung des Abwehrsystems und kann in ihrem Erscheinungsbild sehr variabel sein. Die Erkrankung hat ihren Namen von einem körpereigenen Enzym (Eiweiß), der sogenannten PI3 Kinase delta. Die genetische Veränderung führt zu einer überschießenden Aktivität der PI3 Kinase und das hat eine Störung der Aktivität der Immunzellen zur Folge. Die PI3 Kinase delta hat eine wichtige Funktion in der Informationsweitergabe (Signalweitergabe) in den Abwehrzellen und ist an einer Vielzahl von Abläufen in der Zelle beteiligt. Eine Veränderung der Struktur, der Aktivität, oder der Regulation des Enzyms kann daher diese Zellabläufe stören, was zu verschiedenen Störungen der Gesundheit führt.
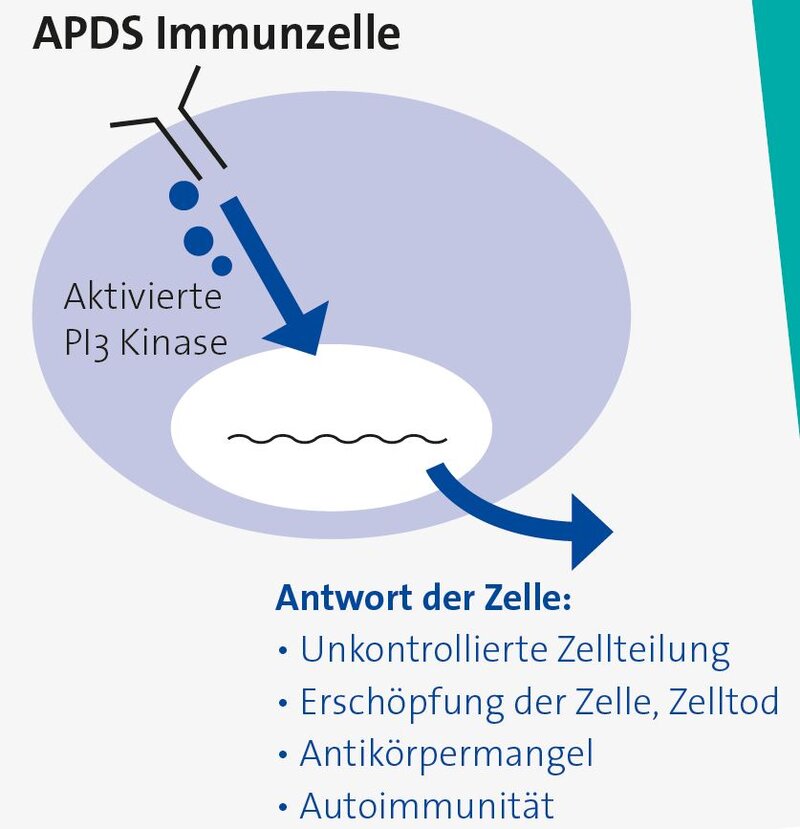
Die verstärkte Aktivität des Enzyms führt dazu, dass Informationen in den Zellen zu lange oder zu stark weitergegeben werden. Den Immunzellen wird so signalisiert, fortlaufend aktiv zu bleiben und sich immer weiter zu vermehren (proliferieren), obwohl der Grund für so eine Vermehrung (z.B. eine Infektion) gar nicht mehr besteht. Es vermehren sich auch Immunzellen, die sich bei normaler Signalleitung gar nicht vermehren, u.a. Zellen, die sich nicht gegen Infektionen, sondern gegen den eigenen Körper richten können. Die ausgeprägte Vermehrung führt mit der Zeit auch dazu, dass Immunzellen vorzeitig altern und absterben. Das führt dann zu einem Mangel an Immunzellen, was wiederum eine Abwehrschwäche nach sich zieht. Die Abwehrschwäche wird dadurch verstärkt, dass Zellen, die zu starke Signale erhalten, ihren normalen Funktionen (z.B. Bildung von Antikörpern) nicht mehr richtig nachkommen können.

„Ich war jedem Infekt hilflos ausgeliefert, jede Krankheit verlief heftig und lange. Vor allem Atemwegsinfekte und Mittelohrentzündungen machten mir zu schaffen, aber auch Magen-Darm-Infektionen.“
Häufig ist der Krankheitsbeginn in der frühen Kindheit, wobei die meisten jungen Patienten mit immer wiederkehrenden Infektionen auffallen. Die Art der auftretenden Symptome, aber auch die Schwere der Beschwerden können sich von Patient zu Patient stark unterscheiden. Auch der Beginn der ersten Symptome ist unterschiedlich, manche Patienten zeigen erst als Jugendliche oder junge Erwachsene erste Krankheitserscheinungen.
Bei der Mehrheit der Patienten sind bereits ab dem frühen Kindesalter Infektionen der oberen Atemwege zu beobachten. Häufig sind das bakterielle Infektionen der Bronchien und Lunge, aber auch Mittelohrentzündungen (Otitiden) und Nebenhöhlenentzündungen (Sinusitiden) treten gehäuft auf. Die Infektionen verlaufen oft schwerwiegender als beim immungesunden Menschen und können zu Schädigungen an den betroffenen Organen führen, wie z.B. zu Bronchiektasen (Aussackungen der Bronchien in welchen sich dauerhaft Keime ansiedeln können) oder zu einer Hörminderung bei wiederholten Mittelohrentzündungen durch Vernarbungen des Trommelfells. Auch Infektionen mit Viren, wie Varizella Zoster Virus =VZV (Windpocken Erreger), Cytomegalie Virus = CMV oder Epstein Barr Virus = EBV (Verursacher des Pfeifferischen Drüsenfiebers) führen zu schwerer verlaufenden Krankheitsphasen, mit häufig lang bestehender Virenlast im Blut.
Nicht selten kommt es im Verlauf der Erkrankung zu einer Schwellung der Lymphgewebe. Hierzu gehören auch die Mandeln (Tonsillen) und Polypen (Adenoide), die die Infektneigung der Ohren, Nebenhöhlen und Lunge noch verstärken können. Häufig kommt es außerdem zu einer generalisierten Lymphknotenschwellung und zu einer deutlichen Größenzunahme von Leber und Milz. Die Milzvergrößerung kann so ausgeprägt sein, dass das Risiko einer Milzverletzung dadurch deutlich steigt und eine Behandlung der Milzgröße erforderlich macht. Bei einigen Patienten ist diese Vergrößerung der Organe der erste Hinweis auf eine Erkrankung des Abwehrsystems.
Die Fehlsteuerung des Abwehrsystems kann sich auch dadurch äußern, dass sich das Immunsystem gegen den eigenen Körper richtet. Der Körper bildet dann Autoantikörper, d.h. Antikörper die nicht gegen „fremde“ (Infekte), sondern gegen körpereigene Strukturen gerichtet sind. Das kann zu verschiedenen Krankheitsbildern (Autoimmunerkrankungen) führen, wie z.B. zu einer Erniedrigung der Anzahl der Thrombozyten, Erythrozyten oder Leukozyten im Blut (Autoimmunzytopenien), oder zu einer Erkrankung der Schilddrüse (Autoimmunthyreoditis). Auch die Gelenke können davon betroffen sein, wodurch es zu schmerzhaften, entzündlichen Veränderungen, meist der großen Gelenke kommen kann.
Bei einigen Patienten findet man außerdem eine Einwanderung von Immunzellen (Infiltration von Lymphozyten) in unterschiedliche Organe, wie z.B. in die Lunge, in den Verdauungstrakt, die Nieren, die Leber, das Gehirn und das Knochenmark. Dies kann zu einer Entzündung und einer Funktionseinschränkung des jeweiligen Organs führen. Veränderungen der Schleimhäute im Magen-Darmtrakt können so bei einigen Patienten zu dem Beschwerdebild einer chronisch entzündlichen Darmerkrankung führen, mit der Folge von wiederkehrenden Durchfällen und gestörter Verwertung von Nahrungsbestandteilen. Das Bild ist ähnlich wie bei einem Morbus Crohn oder einer Colitis ulzerosa und geht häufig mit einer Gewichtsabnahme oder mangelnden Gedeihen bei jüngeren Patienten einher.
Aufgrund der Funktionen die das Enzym PI3 Kinase delta in den Zellabläufen spielt, besteht auch ein erhöhtes Risiko für die Entstehung von Lymphknotenkrebs (Lymphomen). Das Enzym spielt auch eine (kleinere Rolle) in Zellen, die nicht zum blutbildenden System gehören. Dies erklärt wahrscheinlich, warum manche Patienten eine leichte Entwicklungsverzögerung haben oder diskrete Auffälligkeiten in der Kopf- oder Gesichtsform.
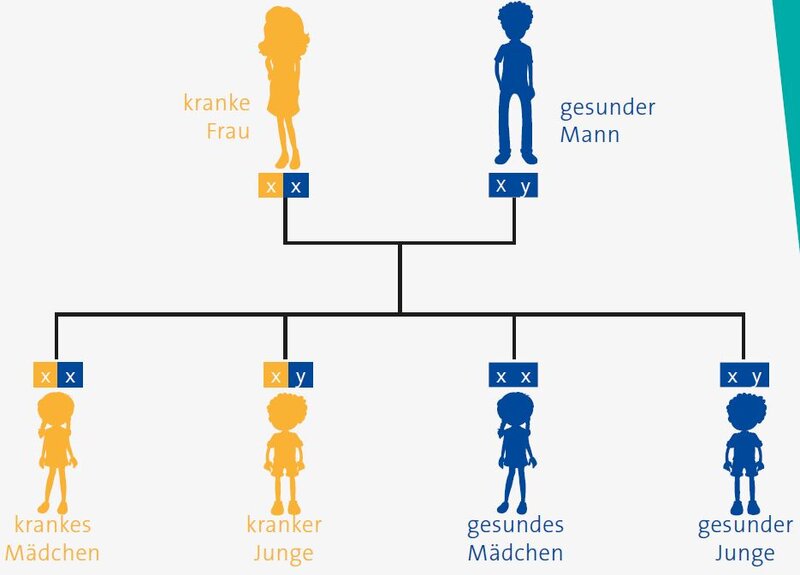
Autosomal-dominanter Erbgang
Wenn eine Erbkrankheit vorliegt, bedeutet das, dass der Patient von Mutter und/oder Vater ein fehlerhaftes Gen geerbt hat. Jeder Mensch besitzt von jedem Gen zwei Stück, eines vom Vater und eines von der Mutter. Für die meisten Erbkrankheiten ist es für den Ausbruch der Erkrankung nötig, dass beide Gene fehlerhaft sind, da ein gesundes Gen in der Regel ausreicht, genügend gesunde Proteine herstellen zu können. Beim APDS, das durch eine „überschießende“ Aktivierung der PI3 Kinase delta bedingt ist, reicht jedoch das Vorliegen eines fehlerhaften Genes, um die Erkrankung hervorzurufen. D.h. wenn ein Elternteil erkrankt ist, liegt die Erkrankungswahrscheinlichkeit bei jedem Kind der Familie bei 50%. Es handelt sich um eine Erkrankung mit einem sogenannten autosomal-dominanten Erbgang.
Die genetische Veränderung kann aber auch bei dem betroffenen Patienten zum ersten Mal auftreten ohne dass Eltern oder weitere Verwandte davon betroffen sind. Auf jeden Fall sollte bei Feststellen der Erkrankung näheren Verwandten, vor allem Eltern und Geschwistern, auch eine genetische Untersuchung angeboten werden.

Gründliche Anamnese und Blutabnahme für weiterführende Laboruntersuchungen.
Um das PI3 Kinase delta Syndrom festzustellen, ist eine sorgfältige körperliche Untersuchung des Patienten, sowie eine genaue Erhebung der Anamnese (Krankheitsgeschichte) erforderlich. Hier ist auch eine genaue Mitbeurteilung der Familiengeschichte von großer Bedeutung, insbesondere mit der Frage nach Infektanfälligkeit, Autoimmunität oder Lymphomerkrankung. Die körperliche Untersuchung und die Krankengeschichte, führen in ihrer spezifischen Ausprägung zunächst zu der Verdachtsdiagnose der APDS Erkrankung.
Im Anschluss sind dann komplizierte Blutuntersuchungen notwendig, die Auskunft über die verbleibende Funktion des Abwehrsystems geben. Ein genaues Muster von immunologischen Auffälligkeiten ist bisher nicht bekannt, jedoch zeigt ein Großteil der Patienten unspezifische Auffälligkeiten bei „Reifungsmarkern“ des Immunsystems und bestimmte Funktionstests sind auffällig. Oft sind mehrere Blutentnahmen notwendig, um die Auffälligkeiten genau zu verstehen.
Manchmal sind zusätzlich auch feingewebliche Untersuchungen z.B. an einem Lymphknoten oder einer Darmgewebsprobe als weiterführende Diagnostik erforderlich. Beweisend ist eine molekulargenetische Untersuchung. Bei dieser Untersuchung wird ein genetischer Test durchgeführt, bei dem eine DNA-Probe (Blutprobe) auf einen Defekt in der Erbinformation für die PI3Kinase delta untersucht wird.
„Die fehlenden Antikörper kann ich mir alleine zu Hause mit einer Pumpe verabreichen, was mir das Leben sehr erleichtert."
Die Therapie ist abhängig von dem Verlauf, der Schwere und dem Zeitpunkt des Auftretens der Erkrankung. Je nach Beschwerden und Komplikationen wird die Therapie individuell gesteuert und angepasst. Leider gibt es noch keine einfache ursächliche Therapie, die die Überaktivität der PI3 Kinase delta auf ein normales Niveau korrigiert. Daher stehen die Behandlung von Infektanfälligkeit und Autoimmunerkrankungen im Vordergrund, sowie der Schutz vor Infektionen.
Die Behandlung von Infektionen bei APDS umfasst eine schnelle und möglichst Erreger gerichtete und ausreichend lange antibiotische Behandlung bei bakteriellen Infektionen. Manchmal ist jedoch auch eine prophylaktische Dauerbehandlung mit Antibiotika notwendig, um den Patienten so möglichst gut vor Infektionen zu schützen. Bei den Patienten, die durch die Erkrankung einen Antikörpermangel entwickeln, besteht die Therapie aus dem Ersatz der fehlenden Antikörper durch Infusionen. Die Antikörper können entweder subkutan als Heimtherapie oder intravenös in der Klinik verabreicht werden. Da die Antikörper nach einer gewissen Zeit im Körper abgebaut werden, müssen die Infusionen regelmäßig wiederholt werden.
Bei Patienten mit einer ausgeprägten Vergrößerung von Milz, Leber oder Lymphknoten kann die Organvergrößerung solche Ausmaße annehmen, dass man mit Einsatz von Medikamenten versuchen muss, die Größe wieder zu reduzieren. Darüber hinaus müssen Autoimmunerkrankungen oder entzündliche Erkrankungen, z.B. in Darm oder Lunge behandelt werden. Oft kann hier Kortison helfen, dieses Medikament ist aber aufgrund seiner Nebenwirkungen für eine längere Therapie in hohen Dosen nicht gut geeignet.
Es werden daher zusätzlich Medikamente eingesetzt, die das überaktive und fehlgesteuerte Immunsystem vorsichtig unterdrücken (Immunsuppressiva). Zu diesen Medikamenten gehört unter anderem das Rapamycin, das bei APDS erfolgreich eingesetzt worden ist.
Der Einsatz von Immunsuppressiva bei Patienten, deren Immunsystem geschwächt ist, ist oft eine schwierige Balance. Die Steuerung der Behandlung sollte daher in die Hände von Ärzten, die Erfahrung in der Betreuung von APDS Patienten haben. Patienten mit einem PI3 Kinase delta Syndrom sollten in einem mit dieser Erkrankung vertrauten Zentrum behandelt werden.

„Zurzeit nehme ich an einer Studie teil, weil ich hoffe, dass mir und anderen Patienten damit geholfen werden kann."
Die Prognose des PI3 Kinase delta Syndroms ist aufgrund der Variabilität der Erkrankung sehr unterschiedlich. Typisch ist ein Verlauf mit wiederholten, z.T. schweren Infektepisoden, zwischen denen aber auch Phasen liegen, in denen der Patient kaum beeinträchtigt ist. Die Autoimmunerscheinungen können akut, aber auch chronisch verlaufen und dann zu dauerhafter Therapie und Reduktion der Lebensqualität führen. Mit Hilfe von Immunglobulinen, prophylaktischen Medikamenten gegen Bakterien und Viren sowie Immunsuppressiva können diese Krankheitszeichen vermindert, aber meist nicht vollständig vermieden werden. Häufig treten nach wiederholten Infektionen chronische Lungenveränderungen auf, die langfristig ein Problem darstellen. Hier ist eine gute Lungenhygiene mit intensiver krankengymnastischer Behandlung ein ganz wichtiges Therapieelement. Grundsätzlich kann die Erkrankung auch durch eine Knochenmarkstransplantation behandelt werden. Diese intensive Therapie hat aber ihre eigenen Risiken, so dass die Entscheidung zu diesem Schritt bei jedem Patienten individuell diskutiert werden muss.
Es werden derzeit neue Medikamente in Studien getestet, die ganz selektiv die Aktivität der PI3 Kinase delta hemmen können. Es gibt berechtigte Hoffnung, dass diese Medikamente in Zukunft sowohl die Infektneigung als auch die Autoimmunerkrankungen bessern können. Allerdings müssen sie lebenslang genommen werden.
Die Wirkung von Impfungen besteht darin, die Bildung von Antikörpern anzuregen. Wenn die Antikörperbildung gestört ist, sind Impfungen nur von begrenztem Nutzen. Bei Patienten mit dem PI3 Kinase delta Syndrom können alle nach STIKO (Ständige Impfkomission) empfohlenen Impfungen mit Todimpfstoffen gefahrlos durchgeführt werden. Die Anwendung von Lebendimpfstoffen (MMR, Rotavirus, nasale Grippeimpfstoffe, Gelbfieber) sollte allerdings nur nach Rücksprache mit einem in der Behandlung von APDS Patienten erfahrenen Arzt erfolgen.
Sind die Patienten mit Antikörperinfusionen (Immunglobulinen) behandelt, erhalten sie sozusagen einen passiven Schutz durch die Antikörper und weitere Standard-Impfungen müssen nicht durchgeführt werden. Dennoch macht es Sinn, die Patienten zusätzlich gegen Influenza (Grippe), FSME (Frühsommermeningoenzephalitis, je Wohnort) und gegen HPV (Humanes Papillom Virus) zu impfen, da diese Antikörper in der Regel nicht in den Präparaten enthalten sind. Eine jährliche Impfung der engen Kontaktpersonen mit dem Influenza-Impfstoff ist zum Schutz des Patienten sinnvoll.

„Wir fühlen uns am CCI bestens aufgehoben und das hilft uns sehr. Klar, ich bin ein krankes Kind, aber ich schaue voller Optimismus in meine Zukunft.“
Kinder mit dem PI3 Kinase delta Syndrom können in der Regel den Kindergarten und die Schule besuchen. Spezielle Isolations- oder Hygienemaßnahmen bringen keinen Vorteil. Bei Überlegungen zur Berufswahl ist eine Beratung sinnvoll.

Regelmäßige Termine in einem Spezialzentrum sind wichtig, um Komplikationen rechtzeitig erkennen und behandeln zu können.
Grundsätzlich kann mit einer Knochenmarkstransplantation das kranke Immunsystem eines APDS Patienten durch ein gesundes ausgetauscht werden. Bei erfolgreicher Transplantation werden alle Risiken durch Infektionen, gestörter Immunregulation und Lymphomentstehung beseitigt. Krankheitserscheinungen des APDS, die nicht mit dem Immunsystem zu tun haben (Kleinwuchs, Entwicklungsverzögerung) können allerdings nicht geheilt werden. Auch Krankheitserscheinungen durch bereits abgelaufene Infektionen (z.B. Narben oder Veränderungen in der Lunge) können nicht mehr beseitigt werden. Der Zeitpunkt für eine Transplantation sollte sorgfältig gewählt werden. Immer müssen dabei Nutzen und Risiken sehr individuell gegeneinander abgewogen werden. Wenn zu lange gewartet wird, können Organschäden bereits ausgeprägt sein und die Aussicht auf eine erfolgreiche Transplantation herabsetzen. Trotz all dieser Schwierigkeiten und der Ernsthaftigkeit der Erkrankung stehen aber viele Therapiemöglichkeiten zur Verfügung und die Gesamtprognose ist besser als bei einer Reihe von anderen Immundefekten.
Stand
September 2017
Hinweis
Wir möchten mit unseren Patientenbroschüren gerne dazu beitragen, dass betroffene Patienten, Eltern und ihr Umfeld die Erkrankung und ihre Behandlung besser verstehen. Die Broschüren sind sorgfältig erstellt und beschreiben die Erkrankung und deren Behandlung. Auch wenn Sie viele Informationen in den Broschüren finden, können diese vorliegenden Informationen keinen Arztbesuch ersetzen.
Autor
Henrike Ritterbusch
+49 (0)761 270-45240
henrike.ritterbusch@uniklinik-freiburg.de
Wissenschaftliche Begleitung
Prof. Dr. Stephan Ehl
+49 (0)761 270-77300
stephan.ehl@uniklinik-freiburg.de
UNIVERSITÄTSKLINIKUM FREIBURG
Centrum für Chronische Immundefizienz
Mathildenstraße 1
79106 Freiburg
www.uniklinik-freiburg.de/cci
