Augenerkrankungen
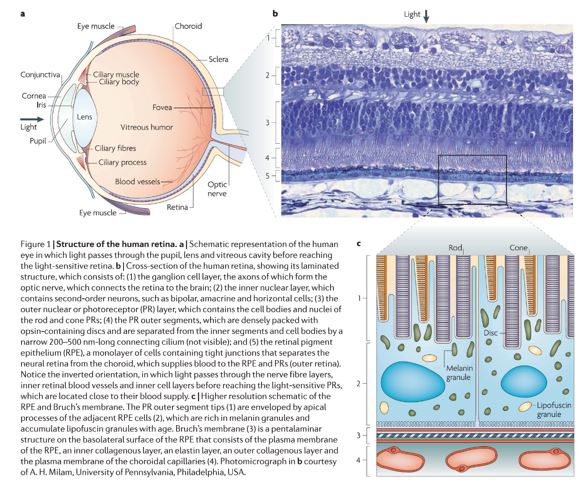
Das menschliche Auge ist ein Sinnesorgan, das auf sichtbares Licht reagiert. Es hat eine annähernd kugelförmige Form, wobei seine äußeren Schichten, wie der äußerste, weiße Teil des Auges (die Sklera) und eine seiner inneren Schichten (die pigmentierte Aderhaut) das Auge im Wesentlichen lichtdicht halten, außer auf der optischen Achse des Auges. Entlang der optischen Achse bestehen die optischen Komponenten aus einer ersten Linse (der Hornhaut - dem klaren Teil des Auges), die den größten Teil des Lichts aus der Außenwelt bündelt; dann einer Öffnung (der Pupille) in einer Blende (der Iris - dem farbigen Teil des Auges, der die Menge des in das Innere des Auges eindringenden Lichts steuert; dann einer weiteren Linse (der Augenlinse), die die restliche Bündelung des Lichts zu Bildern vornimmt; dann einem lichtempfindlichen Teil des Auges (der Netzhaut), auf den die Bilder fallen und verarbeitet werden. Die Netzhaut stellt über den Sehnerv eine Verbindung zum Gehirn her.
Drei Arten von Zellen in der Netzhaut wandeln Lichtenergie in elektrische Energie um, die vom Nervensystem genutzt wird: Stäbchen reagieren auf Licht mit geringer Intensität und tragen zur Wahrnehmung von Schwarz-Weiß-Bildern mit geringer Auflösung bei; Zapfen reagieren auf Licht mit hoher Intensität und tragen zur Wahrnehmung von Farbbildern mit hoher Auflösung bei; und die kürzlich entdeckten lichtempfindlichen Ganglienzellen reagieren auf ein ganzes Spektrum von Lichtintensitäten und tragen dazu bei, die Lichtmenge, die die Netzhaut erreicht, zu regulieren, das Hormon Melatonin zu unterdrücken und den zirkadianen Rhythmus zu steuern.
|
Erbliche Netzhautdystrophien (inherited retinal dystrophies, IRDs) gehören zu einer Gruppe klinisch und genetisch heterogener degenerativer Erkrankungen. Bis heute wurden Mutationen in über 270 Krankheitsgenen mit IRDs in Verbindung gebracht. Eine molekulare Diagnosestellung ist jedoch bei einem Drittel der Patienten noch nicht möglich. Unter dem Begriff Retinopathia pigmentosa (RP) wird eine Gruppe von Erbkrankheiten zusammengefasst, bei denen Anomalien der Photorezeptoren (Stäbchen und Zapfen) oder des retinalen Pigmentepithels zu einem fortschreitenden Sehverlust führen. Die Prävalenz der RP wird mit 1: 4.000 bis 1: 5.000 angegeben. RP kann isoliert oder syndromal auftreten. Die nicht-syndromale RP ist klinisch und genetisch äußerst heterogen und kann autosomal-dominant, autosomal-rezessiv oder X-chromosomal vererbt werden. Die autosomal-dominante RP betrifft etwa 15 bis 25% der Fälle, die autosomal-rezessive RP 5 bis 20% und die X-chromosomale RP 5 bis 15%. Sporadische Fälle sind häufig (40-50%). Der Schweregrad hängt teilweise mit dem Vererbungsmuster zusammen, wobei X-chromosomale Fälle den schwersten Verlauf aufweisen. Die hauptverantwortlichen Gene sind USH2A (autosomal-rezessive RP), RHO (ungefähr 28% der autosomal-dominanten RP) und RPGR (70% der X-chromosomalen RP). |
|
Die Makuladystrophie ist eine seltene Augenerkrankung, die den zentralen Bereich der Netzhaut, die Makula, betrifft. Die Makula ist für das scharfe Sehen verantwortlich, das für detaillierte Aufgaben wie Lesen, Fahren und Erkennen von Gesichtern erforderlich ist. Die Best vitelliforme Makuladystrophie (BVMD) ist eine autosomal-dominante Störung und wird durch Mutationen im BEST1-Gen verursacht. Die Stargardt-Krankheit oder der Fundus flavimaculatus sind progrediente Formen der juvenilen Makuladegeneration mit erheblicher klinischer und genetischer Heterogenität. Die Stargardt-Krankheit wird durch Mutationen in ABCA4 verursacht und autosomal-rezessiv vererbt. Varianten von ELOVL4 sind mit der autosomal-dominanten Stargardt-Krankheit 3 assoziiert. Die weltweite Prävalenz der Stargardt-Krankheit wird auf 1: 8.000 - 1: 10.000 geschätzt. |
|
Die autosomal dominante Optikusatrophie (ADOA) ist klinisch durch eine fortschreitende Abnahme der Sehschärfe ab der frühen Kindheit gekennzeichnet. Die klinische Manifestation kann sehr variabel sein. Die Sehbehinderung ist in der Regel moderat, reicht jedoch von schwerwiegend bis leicht, begleitet von Gesichtsfeldausfällen und Farbsehstörungen. ADOA ist durch den Verlust retinaler Ganglienzellen gekennzeichnet. Ungefähr 80% der familiären und 50% der sporadischen Fälle mit DOA werden durch pathogene Varianten im OPA1-Gens verursacht, welches für ein mitochondriales Innenmembranprotein kodiert. |
|
Die Leber hereditäre Optikusneuropathie (LHON) tritt typischerweise bei jungen Erwachsenen als beidseitiger, schmerzloser, subakuter Sehverlust auf. Die LHON tritt in der Regel im zweiten und dritten Lebensjahrzehnt auf, wobei 90 % der Betroffenen ihr Sehvermögen vor dem Alter von 50 Jahren verlieren. Männer sind vier- bis fünfmal häufiger betroffen als Frauen, aber weder das Geschlecht noch der Mutationsstatus haben einen signifikanten Einfluss auf den Zeitpunkt und die Schwere des ersten Sehverlusts. Neurologische Anomalien wie posturaler Tremor, periphere Neuropathie, unspezifische Myopathie und Bewegungsstörungen sind bei Personen mit LHON häufiger als in der Allgemeinbevölkerung. LHON-verursachende pathogene mtDNA-Varianten sind durch eine reduzierte Penetranz gekennzeichnet. Die Penetranz in verschiedenen Zweigen derselben Familie und zwischen Familien mit derselben LHON-verursachenden mtDNA-Variante kann stark variieren. |
|
Unter Hornhautdystrophien versteht man im weitesten Sinne vererbte Erkrankungen, die eine beliebige Schicht der Hornhaut betreffen und in der Regel fortschreitend sind, bilateral auftreten und keine systemischen Auswirkungen haben. Hornhautdystrophien werden in vier Klassen eingeteilt: epitheliale und subepitheliale Dystrophien, epithelial-stromale TGFBI-Dystrophien, stromale Dystrophien und endotheliale Dystrophien. Während einige Hornhautdystrophien im Laufe des Lebens des Patienten nur wenige oder leichte Symptome und Morbidität verursachen, können andere fortschreiten und schließlich zu erheblichen Seh- und Augenstörungen führen, die einen medizinischen oder chirurgischen Eingriff erfordern. Bei Patienten mit fortgeschrittenen Hornhautdystrophien kann eine Hornhauttransplantation, entweder mit Spendergewebe in voller Dicke oder in Teildicke, angezeigt sein. |